Прежде всего, необходимо располагать программным обеспечением. В этой заметке разобраны действия с интерфейсом пакета визуализации молекулярной динамики Visual Molecular Dynamics (VMD) позволяющего отображать расчеты, выполненные с помощью рассчетного кода Nanoscale Molecular Dynamics (NAMD). Оба эти пакета нахожятся в свободном доступе и скачиваются с сайта разработчика - исследователей из Иллинойского университета Урбана-Шампейн http://www.ks.uiuc.edu/. К каждому пакету имеются руковдства (tutorials), снабженные набором файлов с примерами построений. Поэтому по ходу описания будут встречаться ссылки на эти документы.
Рассмотрим последовательность действий с нуля.
0) Нахождение pdb файла всеатомной структуры
Предположим, вам известно название соединения. Первым делом ищется файл (любого формата), содержащий информацию о положении атомов молекулы в лабораторной системе координат. Информация по известным белкам и белок-лигандным комплексам находится на ресурсе Protein Data Bank https://www.rcsb.org/pdb/home/home.do. Структуры низкомолекулярных соединений таких, как: жирные кислоты, моно- и олиго- сахариды, неорганические соединения, добываются с баз данных типа PubChem https://pubchem.ncbi.nlm.nih.gov/. Структуры белков, как правило можно найти в pdb формате. Остальных видов веществ - необязательно. Файл координат обязательно нужно пересохранять в pdb формат. Для этого структуру нужно загрузить в vmd (2), выделить соответствующую молекулу из списка щелчком мыши, выбрать в меню File опцию "Save Soordinates".
VMD работает с широким спектром расширений, но у меня как-то не захотел открывать xml файл. В таких случаях работает конвертация в любой другой читаемый формат. Помочь в этом может программа OpenBabel.
1) Создание psf всеатомной структуры
Однако одной только информации о расположении молекулы в пространстве будет недостаточно для запуска молекулярной динамики. Положение объекта в следующий момент времени находится путем решения системы дифференциальных уравнений - уравнений Лагранжа, а значит необходимо знать величину электрического заряда и массу каждого отдельного атома. Во внимание принимается и наличие химической связи между атомами. Сгенерировать файл такого типа - psf (Protein Structure File) можно двумя путями (1). В главном окне (Main Window) программы есть вкладка "Расширения" (Extensions), в ней нужно выбрать Modeling и далее Automatic PSF Builder. Всплывет окно с настройками по умолчанию, которые пока трогать не нужно.
Второй способ - задавать команды на исполнение через консоль, для этого нужно перейти: Extensions -> Tk Console где указать путь к исполняемому файлу. Наполнение которго задается руками, расширение ставится pgn. Минимальный набор команд pgn-файла:
запуск модуля, ответственного за генерацию:
package require psfgen
указание на файл топологии (конкретно этот файл есть в библиотеке VMD и содержит информацию о структуре некоторых аминокислотных остатков и липидных молекул):
topology top_all27_prot_lipid.inp
запись всех атомов, содержащиеся в молекуле из your_molecule.pdb в новый сегмент U:
segment U {pdb your_molecule.pdb}
перенос из файла координат атомов, скопированных в сегмент U:
coordpdb your_molecule.pdb U
добавление в структуру сегмента U недостающих атомов водорода (на основании информации из файла топологии):
guesscoord
запись файла координат с отредактированной структурой:
writepdb your_molecule_new.pdb
запись psf для отредактированной структуры - your_molecule_new.pdb:
writepsf your_molecule_new.psf
Далее следует запуск файла:
-dispdev text -e ubq.pgn
Если всё пошло хорошо, то в текущую директорию сохранится два файла - psf, ради которого все затевалось и дублированный pdb.
Вновь сгенерированный pdb может отличаться от исходного, если, например вы изначально скачали файл облегченной структуры не содержащей атомов водорода. Тогда программа на основании своих данных достраивает атомы в тех местах, где число связей меньше, чем позволяет валентность.
2) Создание pdb, par, top файлов грубозернистой модели структуры
После получения pdb и psf файлов для атомной структуры молекулы можно приступать к созданию грубозернистой модели. В той же вкладке Extensions главного окна VMD выбираем Modeling -> CG Builder. Всплывет промежуточное окошко, в котором нужно выбрать тот способ создания модели, который вам нужен. Способ номер один (3). Дробить молекулу на части можно с учетом функциональных групп, ставя каждой в соответствие небольшую бусину, наследующую суммарную массу и заряд всей группы атомов. Такое разбиение получило название "Грубозернистое построение на основе функциональных групп" (Residue-Based Coarse Graning - RBCG). Способ номер два (4). При моделировании систем, размеры которых на порядки превышают размер отдельной молекулы, пользуются "Грубозернистым построением на основе формы исходной молекулы", или (Shape-Based Coarse Graning - SBCG). Здесь становится возможным заменять какое угодно количество атомов на бусину. Имеет смысл выбирать масштаб разбиения исходя из требований точности эксперимента, так, чтобы хотя бы приблизительно сохранить знак плотности распределения заряда на поверхности (помочь в этом может сравнение разукрашенных по величине плотности заряда всеатомной и грубозернистой модели молекулы. Вкладка Graphics -> Representations -> Coloring Method -> Charge).
Разберем здесь случай SBCG-разбиения (для RBCG- последовательность действий будет та же). Во всплывающем окне выбираем Create SBCG Model, жмем Next, переходим в окно билдера где настраиваем параметры. Интерес представляют Learning Parameters. Обучающими они названы потому, что положение бусин определяется сетью не сразу, а за несколько итераций. Почитать про алгоритм вычисления координат бусин можно здесь  MART93B (pdf, 625КБ). Количество бусин нужно указать то самое, на которое вы хотите разбить молекулу. В определении количества "химических" связей между готовыми бусинами есть некоторый произвол. Галка на Determine Bonds From All Atom гарантированно скопирует связи со всеатомного представления молекулы на масштаб бусин. Однако радиус связанности бусин можно задавать руками (в Ангстремах) установив отметку возле Provide Bond Cutoff. Так появляется возможность варьировать подвижность внутренних частей молекулы - представлять её абсолютно твердым телом (при задании большего радиуса), либо устранять некоторые связи, присутствующие во всеатомной структуре (при задании меньшего радиуса). После нажатия Build Coarse Grain Model статус процесса начнет отображаться в нижней части окошка. По окончании вычислений будут сгенерированы четыре файла: файл бусинных координат (pdb), файлы топологии (top) и силовых параметров (par) новой модели (о типах файлов см. (1)); файл исходной структуры с пометкой "reference", чтобы никто не забыл, с какого оригинала была слеплена грубозернистая модель.
MART93B (pdf, 625КБ). Количество бусин нужно указать то самое, на которое вы хотите разбить молекулу. В определении количества "химических" связей между готовыми бусинами есть некоторый произвол. Галка на Determine Bonds From All Atom гарантированно скопирует связи со всеатомного представления молекулы на масштаб бусин. Однако радиус связанности бусин можно задавать руками (в Ангстремах) установив отметку возле Provide Bond Cutoff. Так появляется возможность варьировать подвижность внутренних частей молекулы - представлять её абсолютно твердым телом (при задании большего радиуса), либо устранять некоторые связи, присутствующие во всеатомной структуре (при задании меньшего радиуса). После нажатия Build Coarse Grain Model статус процесса начнет отображаться в нижней части окошка. По окончании вычислений будут сгенерированы четыре файла: файл бусинных координат (pdb), файлы топологии (top) и силовых параметров (par) новой модели (о типах файлов см. (1)); файл исходной структуры с пометкой "reference", чтобы никто не забыл, с какого оригинала была слеплена грубозернистая модель.
3) Создание psf грубозернистой модели структуры
Теперь необходимо создать psf для сгенерированной грубозернистой модели структуры (1). При использовании графического интерфейса PSF Builder к списку файлов топологии добавляется файл топологии грубозернистой модели молекулы. При работе с консолью содержание pgn файла выглядит так:
package require psfgen
topology cg_molecule.top
segment F {pdb cg_molecule.pdb}
coordpdb cg_molecule.pdb F
guesscoord
writepdb cg_molecule_rewritten.pdb
writepsf cg_molecule.psf
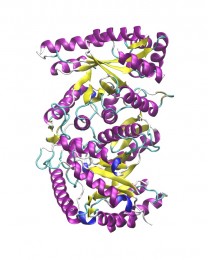
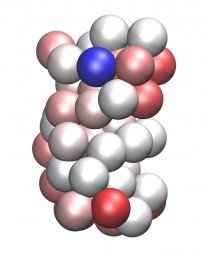
Вот как выглядит одна и та же молекула - бактериальная люцифераза - во всеатомном (верхняя картинка) и в грубозернистом (SBCG) представлении:
4) Запуск молекулярной динамики
Молекулу можно поместить в растворитель, посмотреть, как она будет двигаться. Для этого в файле с расширением conf указываются необходимые параметры. Удобно взять файл конигурации из архива, прилагающегося к руководству по составлению sbcg-модели, переписав лишь путь к pdb, psf и par файлам структуры, и выставив нужное число шагов минимизации энергии и дальнейшей динамики.
Ещё нужно не забыть указать координаты геометрического центра ячейки, содержащей растворяемую структуру.
cellOrigin 225.0 50.0 0.0 // координаты указаны по осям {x, y, z};
Наклон плоскостей параллелограмма сольватоционного бокса задается следующими векторами, сумма которых определяет верхний угол ячейки:
cellBasisVector1 1000.0 0. 0.
cellBasisVector2 0. 110.0 0.
cellBasisVector3 0. 0. 500.0
Для того, чтобы задавать координаты параллелограмма, куда помещается молекула, нужно знать расположение самой структуры.
Определить их можно задав в консоли команды:
set everyone [atomselect top all]
где atomselect all — команда выбора всех атомов молекулы под порядковым номером, присвоенным ей при загрузке;
top — указывается вместо номера для верхней в списке молекулы;
set everyone — команда присвоения в переменную с названием everyone набора атомов, указанных в скобках;
measure minmax $everyone
определение координат концов диагонали прямоугольного параллелограмма, в который вписана молекула;
Рассчитать геометрический центр ячейки можно руками или с помощью команды:
measure center $everyone
*** Обратите внимание, что в конфиге для грубозернистой модели присутствует блок "Implicit Solvent" (Неявный растворитель). Это сделано для того, чтобы не учитывать отдельно каждую молекулу воды (размер и масса которых может быть на порядки меньше параметров бусины) (вариант Explicit Solvent - модели явного растворителя), а работать с некоторой экранирующей заряд средой. По сути, переход от микропараметров среды (скорость и расположение молекул растворителя) к макропараметрам (постоянная экранирования).
Остается запустить файл конфигурации (1)
namd2 cg_mol_solv.conf > cg_mol_solv.log &
где namd2 — указание на файл, выполняющий запуск NAMD-а; при необходимости прописывается полный путь к файлу;
cg_mol_solv.conf — файл с настройками,
> cg_mol_solv.log — журналирование статуса промежуточных процессов в файл cg_mol_solv.log;
По окончании расчета текущая папка пополнятеся 9 файлами, один из которых - журнальный. Для просмотра анимации необходимо загрузить файлы с расширением dcd и старый psf.
5) Дополнительные действия
Гораздо интереснее работать со сложными системами. Подробнее (5). Например, для генерации липидных мембран предусмотрен отдельный билдер: Extensions -> Modeling -> Membrane Builder. Формируется липидный бислой с заданным составом http://www.ks.uiuc.edu/Research/vmd/plugins/membrane/.
Для добавления в систему дополнительных молекул ранее добавленные можно сдвигать
$everyone moveby {37.0 85.5 24.7} // вектор сдвига центра молекулы
И записывать ранее отдельные молекулы в один pdb-файл.
Литература (http://www.ks.uiuc.edu/Training/Tutorials)
1. NAMD tutorial for Unix/Max
2. VMD User’s Guide (уже содержится в папке программы)
3. Residue-Based Coarse Graining
4. Shape-Based Coarse Graining
5. Membrane Proteins Tutorial